Processing a High-Resolution Cryo-Tilt Series with Fiducials
(IMOD 5.1)
University of Colorado,
Boulder
This is a guided introduction to generating a tomogram from a
cryo-tilt series to be used for high-resolution subvolume averaging, with gold beads as fiducial markers for alignment. It
presents the most important concepts and details and provides brief explanations
of some points. For more details, consult the Tomography Guide, which you
can open from the Help menu in Etomo. It is also advisable to read through
relevant sections of the Tomography Guide before trying to process your own tilt
series. If you have never processed a tilt series in Etomo before, you may want
to do the dual-axis tutorial before this
example, especially if you have difficulty following the steps below without
shots of how the screen should appear.
The example data set is from the study of HIV virus-like particles in Schur et
al., 2016, Science 353:506-598, based on the data set TS_43 from the
EMPIAR-10164 deposition. The tilt series was produced by aligning
the super-resolution K2 frames with Alignframes, using the command
alignframes -pair 8 -bin -1,2 -mdoc TS_43.mrc.mdoc TS43.mrc -adj -dty 4 -norm -ref 5 -gpu 0.
This data set is set up to be used as an example for
measuring CTF, and the reconstruction will be done in a subdirectory.
First, a few points on conventions: labels in the Etomo or 3dmod interface are shown
in Bold, and entries in fields are shown in italics. For
mouse operations in the Zap window in 3dmod, the buttons are referred to as
"first", "second", and "third" because the buttons can be remapped in 3dmod.
If you have not changed the mapping, this corresponds to left, middle, and
right; otherwise, it refers to whatever you have chosen to be the first, second,
and third buttons.
Getting started:
- If you did not already do the tutorial
on Finding Defocus with Ctfplotter:
-
Download the sample data set from our web site.
- Move the data set file "TS43-CTF-Data.tar.bz2" to the directory where you want to
work on it. Its contents will unpack into a subdirectory named "TS43-ctf".
- cd to the directory with the downloaded file
- Enter the command:
imoduntar TS43-CTF-Data.tar.bz2
or, anywhere except on Windows without Cygwin, you can use
tar -xjf TS43-CTF-Data.tar.bz2
- Enter the data set directory with:
cd TS43-ctf
- Enter this command to move needed files to a subdirectory:
subm setupRecon
-
Enter the directory for reconstruction with
cd reconTS43
- Start the Etomo interface with:
etomo
Tomogram setup:
In this initial step, we define some features of the data set and create the
files needed for processing.
-
Press Build Tomogram in the Etomo "Front Page"
-
Press the yellow file chooser icon on the Datatset name line and select the stack file,
TS43.mrc.
-
For SystemTemplate, select
cryoAccurate.adoc. This will set numerous parameters appropriately for cryo
reconstructions in general, and a few for high-resolution reconstructions. You should generally select a template.
-
Press Scan Header to retrieve the pixel size and rotation angle of
the tilt axis from the image file.
-
Enter 10 for Fiducial diameter
-
Select Parallel Processing if you have more than one CPU core.
-
Select Graphics card processing if you have an Nvidia card with a GPU that can be used with IMOD.
-
Check Series was and select dose-symmetric.;
the starting angle here was 0 degrees, but if it was not, you would have to fill
in the from field. More recent tilt series from SerialEM
have an entry in the file header specifying this and Etomo would set these
fields correctly.
- Press View Raw Stack and scroll through the images to see that they are
not very well-aligned. Notice the 4 contrast control sliders, which are
present because the data have been loaded as integers. The top two sliders
select a window of the data range that is mapped to bytes for display. Sometimes these two
sliders are much closer together than here, which means the image data occupy a
very small fraction of the full data range, with the rest of the data range taken
up by image artifacts. In general, if you see bad views that you know
that you want to exclude from alignment and reconstruction, you can list
these in the Exclude views box.
- Press Create Com Scripts
Pre-processing:
This step is
often needed to remove artifacts in the images, generally due to "hot pixels" in
a direct detector camera. These artifacts will produce streaks in a
reconstruction and can also make it harder to see the image features, which have
a much smaller dynamic range than the artifacts.
-
Press Show Min/Max for Raw Stack to
see the range of the data; both a plot of minimum and maximum values and a table
with more detailed information will open. Some of the views have maximum
values that are substantially higher than for adjacent views.
-
Press Create Fixed Stack to run the program that finds and erases
artifacts.
-
Press View Fixed Stack. Now there are only 2 contrast sliders because data
can be loaded as bytes after removing most of the artifacts.
-
Press Show Min/Max for Fixed Stack to
see the new range of data.
The outlying maximum values are gone.
- Press Use Fixed Stack.
- Press Done to advance to the next step.
Coarse Alignment
In this step, we use image cross-correlation to align successive images, which
makes it easier to track fiducial markers.
-
Press Calculate Cross-Correlation
-
When done, press Generate Coarse Aligned Stack to
generate an unbinned stack. In the past it was customory to select a high
binning at this stage to bring the beads into a standard size range, but this
became unnecessary in IMOD 4.11, and fiducial positions will usually be found more accurately
with images that are unbinned or binned by only 2.
-
When done, press View Aligned Stack in
3dmod. Scroll through the images
to see that they now look reasonably well aligned.
-
Press Done to go on.
Fiducial Model Generation:
In this step, the positions of selected gold markers are found on each
image, or as many of them as possible, which allows a more accurate alignment to be obtained.
-
Use the default Make seed and track and Generate seed model
automatically options. Making a model manually is covered in other
tutorials.
-
Enter 20 in the Total number box.
-
The option to Use boundary model is available in cases where you do not
want fiducials outside of a defined area. In cryoET, it is usually
desirable to avoid using beads over the carbon, if possible.
-
Press Generate Seed Model.
-
When done, the project log reports that only 16 beads were selected, with most
of the rest being identified as clustered, and a few as elongated. The
program that picks good seed points considers beads to be clustered if they are
in danger of overlapping higher in the tilt range. Press Open Seed
Model to see what it selected.
-
You should see a fairly good (i.e., even) distribution of modeled beads, except that an
isolated bead in the upper right has not been picked because it was considered
too elongated. The main concern with elongated beads is that they may appear that
way because they closely overlap another bead or some strong density, making the
measured center position wander away from the true bead center over parts of the
tilt range. That is clearly not the case here, and the elongation is
minor, so it is worth adding the bead. If this were a more widespread
problem, we could turn on Advanced mode and set an option to accept some
elongated bead. Here it is quicker just to add the bead manually, as
described next.
-
Turn on Automatic new contour in the Bead Fixer window if it is not on already. A contour is
a set of connected points, in this case representing the location of a
particular gold bead throughout the tilt series. Initially, (i.e.
immediately after seeding), each contour should contain just a single point
locating the bead in some view. Beadtrack will subsequently add locations
on other views to each contour.
-
Place the cursor
over the bead and add the point with the second mouse
button. It will be automatically centered. (The Autocenter option is
turned on by Etomo in seeding mode.)
-
Press the S key to save the model.
-
Switch to the Track Beads tab and press Track Seed Model.
-
The Project Log window shows the number of missing points when done. It
may be 0.
-
Press Fix Fiducial Model, which will load the tracked model into 3dmod and
switch the Bead Fixer to Fill gaps mode. You can scroll through the
views and also press the V key to see the tracks of the beads in 3D.
If there are missing points, you can now use the Bead Fixer to step from one gap
to the next (using the space bar as a hot key) and add a point, if appropriate.
If you add points, either be sure to center them well or turn on the Autocenter
option, which was turned off for this mode because it does not work well in many
cases where a gap was left. A bead does not need to be
marked on every view, and you should not add a point if the bead's position is
not clear. If you add points, be sure to save the model with the S
key.
-
Another way to complete the model is to press Track with Fiducial Model as
Seed. It is a good idea to look at the model in 3D first and make sure
there are not deviant points at the ends of contours that should be
fixed first.
-
Press Done to go on.
Fine Alignment:
Next, the bead positions are fit to a mathematical model of specimen movements.
The model predicts a position for each bead on each view, and the mean distance
between the predicted and actual positions is referred to as the "mean residual
error". By obtaining a solution with some points left out, a
different error can be computed between the predicted and actual positions of
points not included in the fit, the "leave-out" error. It
requires many alignment runs with different points left out to obtain a reliable
estimate of the leave-out error. Since this error is an estimate of how well the
solution generally predicts the positions of structures other than the beads
that happen to be included in the fit, it is a better measure of the quality of
a solution than the mean residual. The main problem with the mean residual
is that solving for more variables will always give a better fit to the points,
but using too many variables will fit excessively to the random errors in the
positions and do a worse job of predicting the positions of other points in the
specimen. You should always use the leave-out error to assess the value of any
change in the alignment parameters. However, either of these errors will let you find and correct badly modeled points.
We will go through a preliminary adjustment of parameters to improve the fit,
then fix some of the points with the largest errors, then do a final
optimization of the parameters.
Initial parameter adjustment
The reason to do at least some adjustment of parameters at the start is to
minimize the number of points that have large residuals because the model is not
complex enough to fit them, so that we can focus on finding points that are not
well-centered.
-
Notice that Do not sort fiducials into 2 surfaces for analysis has been
selected because this option is set in the cryo templates.
-
Press Compute Alignment. When it is done, the Residual error
mean and the Global leave-out error will appear in the Project Log (e.g., 0.120
and 0.1454 nm).
-
Press View 3D Model to open the model of solved positions in 3D and
assess whether to solve for distortion (stretching and skew). Press the R
key in the Model View window for a side-view of the model. Press the
middle mouse button and move the mouse up and down, to rotate the model around
the X axis, and try to make the upper row of points be as planar as possible.
Note that a few of these points deviate somewhat from the plane that the others
lie in. Now move the mouse up enough to rotate the model back by 90
degrees to the top view, and watch where the 4 points in the lower row end up.
Two of them are neighbors but the other two are fairly well separated.
-
To solve for distortion, fiducials should be well-distributed in Z: not all on
one plane, more than a few at different Z heights, and the ones at different
heights distributed over the area. This is not often the case for cryo data
sets, and when these conditions are not met, the distortion and tilt angle can
play off against each other so that an infinity of solutions fit the points
nearly equally well. This set is right on the edge, with only 4 of 16 on the
bottom and somewhat well-distributed, but the raggedness of the points in the
upper row should help to prevent tilt angle and distortion from being redundant
variables.
-
Switch to the Global Variables tab and select Full solution in
the Distortion Solution Type box.
Press Compute Alignment. The leave-out error drops quite a bit (e.g., from
0.1454 to 0.1360).
-
When solving for distortion, it is a good idea to check whether reasonable
results have been obtained.
Right-click in Etomo over the Fine Alignment panel and select Align
log file. It opens with the Errors tab, which has an error summary. Switch to the
Solution tab to see the alignment parameters. The X-stretch and skew are
in the columns "dmag" and "skew". When there are relatively few fiducials
distributed in Z, as here, the distortion solution should not be trusted if the
X-stretch exceeds ~0.025 or the skew exceeds ~2 degrees. The values here
are fine.
-
Another way to look at various solution components is to right click over the
Fine Alignment panel and select one of the Plot options, in this
case the X-stretch one.
-
An apparent distortion of the specimen could also result from the beam not being
perpendicular to the tilt axis, which is referred to as beam tilt. It is
worth checking whether this single variable can give a nearly comparable result.
To do so, select Disabled again for the distortion solution
because beam tilt and distortion cannot be solved together - they have much too
similar an effect on bead positions. Press the A
(advanced mode) button
on the Single Variables: Beam Tilt, X Tilt, Projection Stretch
box and select Solve for beam tilt. Compute the
alignment. The leave-out error shows a drop almost as big as with
distortion (e.g., to 0.1373 nm, 86% of the reduction with distortion).
This shows the advantage of solving for beam tilt when the distribution of
fiducials does not allow solving for distortion, but we might as well stay with
the variables that give the lowest leave-out error. Return the selections
to No beam Tilt and Full solution.
-
Another single variable worth checking is Solve for single stretch
during projection, which accounts for anisotropic magnification as
opposed to stretching of the specimen. Check this and compute alignment
again. There is another drop in leave-out error (e.g., from 0.1360 to 0.1338).
Correcting model points
In some situations, the checking of model points can be skipped by using a
method called "robust fitting", which automatically gives less weight
to or even
eliminates the points most likely to be at incorrect positions. However,
this method often does not help much (as we will see), and some manual checking of
positions is appropriate when doing high-resolution work.
-
Press View/Edit Fiducial Model. The Bead Fixer will open in (or
be switched to) Fix big
residuals
mode and read in the log from alignment. Set the zoom of the Zap window so
that you can easily judge whether a green circle is centered on a bead, and so
that small changes in position can be done with the mouse. It is also helpful to switch the Zap window to keeping the current
model point centered, by pressing the concentric squares in the toolbar.
-
Pressing Go to Next Big Residual or the single quote (') key will
move to the next potentially problematic point, sorted in order of decreasing
residual magnitude. If a
point is not centered on the gold bead, you can move it to the correct position
by clicking with the third mouse button. The red arrow points to the
position predicted by the alignment solution; it is not based on any knowledge
of what is in the image. This position may not be
correct, but if it is, you can move the point to that position with
Move Point by Residual or the semicolon (;) key.
-
After fixing all the points offered, press Save & Run Tiltalign.
It will probably offer some new points. You can repeat this process until
either no points are left, or the changes in position become imperceptible.
You should be able to get the residual error down a few percent.
-
Sometimes all the biggest residuals are at high tilt, yet all the points appear
well-centered there. This happens when the alignment model does not fit
the positions well enough at high tilt. That is not the case here, but when it
happens, it means that some points at lower tilt angles may be less
well-centered but still have lower residuals. To find these points, you
can switch the Residual Reporting to be Relative to Neighboring Views
and rerun the alignment.
Optimizing parameter settings
The goal of this step is to avoid overfitting to the random position errors by
reducing the number of variables being solved for. It is important but simple to do.
-
First, return to the General tab and turn on Do robust fitting
to see if that will help at this point.
-
Press Run Cross-Validation.
-
When it is done, you should see in the project log that it turned off robust
fitting because it gave negative benefit, and turned on grouping of rotations
and magnifications, with a 2.5% reduction in leave-out error.
-
Press Done to go on.
Tomogram Positioning
The goal of this step is to set angles and an offset in Z so
that the specimen is level in the X-Z plane and centered in Z in the computed volume, thus minimizing the
computational effort.
-
Set Sample tomogram thickness to 1800.
-
Select Use whole tomogram and set Binning to
8
-
Press Create Whole Tomogram to build a whole, binned-down tomogram.
-
Press Create Boundary Model to open the tomogram
-
Open an XYZ window with Image-XYZ. Set
Sum in the toolbar to 50, which makes it average 50 slices.
-
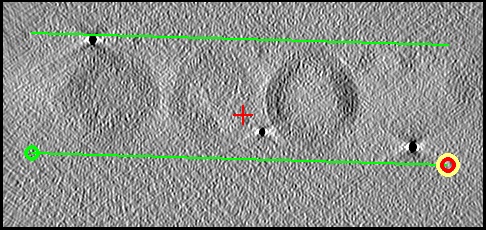
The goal is to draw a pair of lines enclosing the sample at 3
or 4 different Y
locations in the tomogram.
-
Use the Y slider in the toolbar to scroll through Y and get a sense of
how the boundaries change and where the VLPs are well-defined. There are
good locations near Y positions 28, 141, 286, and 346. Since none
of these are close to the middle in Y, and there is no clear boundary to the specimen except
the tops and bottoms of th VLPs, it is preferable to do all 4 positions
instead of just 3.
- Go to a good slice
at one of these locations and draw two lines bounding the upper and lower
surfaces of the sample in the Slicer XZ view. 3dmod will automatically start
a new contour after two points. Move the current point with the third
mouse button; use the first mouse button to select a new current point. See the
image below.
-
Do the same at the three other locations. Your final model should
contain 8 contours with 2 points each.
-
Save the model with the S key.
-
Set Added border thickness to 25. This entry, in
unbinned pixels,
adds a border to the region defined by the drawn lines. Given how binned-down
the tomogram was,how thick it is going to be, and how imprecise the borders are,
more than the default is appropriate.
-
The option to Keep X-axis tilt at zero was set by the
"cryoAccurate.adoc" template. This means that no X-axis tilt will be
applied to make the tomogram level in the Y direction. The two main
reasons for doing this for high-resolution data are to keep the missing wedge at
a standard orientation in the tomogram and in any subvolumes being averaged, and
to avoid an extra interpolation step when using the simplest kind of alignments
(which we are not, since we included the distortion solution).
-
Press Compute Z Shift & Pitch Angles. The computed angle offset, Z shift, and tomogram thickness are shown; these will give the thinnest, most level
tomogram containing the area you outlined with X-axis tilt kept at zero.
-
Press Create Final Alignment, which reruns the fine
alignment.
-
Press Done to go on.
Final Aligned Stack Creation and CTF Correction:
-
Press Create Full Aligned Stack.
-
Switch to Correct CTF tab. Set Voltage (KV) to 300
and Spherical Aberration (mm) to 2.7.
-
The tilt series in EMPIAR 10164 were taken at nominal defocuses range from ~1.5
to 4 microns. We will pretend that we do not know what defocus this particular set
is supposed to have; set Defocus range to scan (low, high in microns) to
1,6.
-
Clear out the text box for the Expected defocus. When scanning a
defocus range, this entry is optional to provide additional guidance.
-
Processing this set with Ctfplotter is explained in detail in the first section
of Finding Defocus with Ctfplotter for Four Example
Tilt Series. You can either go through that example now or
follow the rest of the steps in this section to determine the CTF parameters
quickly.
-
Press Run CTF Plotter. Ctfplotter loads some images and
then displays the power spectrum. In brief, the X axis is spatial frequency in
cycles per pixel, thus running from 0 to 0.5. The Y axis is the
logarithm of a rotationally averaged power spectrum, summed from
overlapping subareas referred to as tiles. The red or magenta curve
is the actual power spectrum; the green curve is a curve fit to it
based on equation that behaves like the CTF. The defocus value that
results from this fitting is shown in the top line after D: In this case, scanning
found the correct value, ~2.5 micron.
- Check Find astigmatism in the Fitting Params dialog. It finds a small astigmatism
value, shown in the top line.
- Uncheck Show wedge fits in that section of that dialog. (In practice, you would
probably not do this so soon.)
- Press the Autotune button in the upper left of the plot window. The
program finds a good set of parameters for sampling the power spectrum and fitting to it.
- Press the Assess button on the right end of the line under Tile & wedge parameters
in the Angle Range & Tile Selection dialog. It finds a Tilt offset of 0.5 degrees.
- Check Fit each view separately in the Autofit section of the dialog.
- Press Autofit All Views
- When it is done, you can press Graph Values at the bottom of the dialog to see defocus and astigmatism
plotted versus tilt angle.
- Press Save to File at the bottom of the dialog and exit Ctfplotter.
- You do not need to make a CTF-corrected stack because you are going to do a
3D CTF correction during reconstruction. No parameter
changes are needed in the CTF Correction section.
Gold Erasing:
Gold beads are by far the densest items in cryo-reconstructions and they
cast artifactual rays that are about as dense as the biological features.
To minimize this effect, it is often desirable to remove the beads from the
projection images before reconstruction.
- To erase gold, select the Erase Gold tab.
- Select Use findbeads3d to find the locations of the gold in a
binned-down tomogram and project their positions onto the tilt series images.
- Etomo has set the Thickness and Added Z Shift
to values that will produce a tomogram with all of the modeled fiducials within
the volume. Such a tomogram will contain all gold beads only if the modeled ones
span the full range of the rest of the beads in Z. This is generally the case for plastic
sections where beads are confined to one or two surfaces, but may not be the
case for a cryosample. To improve your chances of getting all the beads on
the first try, set the Thickness at least as big as the value found in positioning.
-
Notice that the Aligned image
stack binning is set to 12. This value was set by Etomo to bring the bead
size down to about 5 pixels.
-
Press Align and Build Tomogram.
-
When it is done, select Store only points above threshold and press
Run Findbeads3d. This will find the beads in the tomogram as well as
it can, and only put the points into the model that have "peak strengths" above
what it thinks is the best threshold for discriminating beads from non-beads.
-
When it is done, press View 3D Model on Tomogram. The Bead Fixer
opens with some special controls for adjusting the threshold peak strength
(useful if we had stored additional below-threshold points in the model.)
-
You can scroll through the images to see how well it found and localized the
points, or step though the contours with the C or Shift C hot key to check the
centering. It should be fine, but if you get an extra point not on a bead,
you can delete it with the Shift D hot key when it is the current contour. Close 3dmod.
-
Press Model then View 2D Model on Aligned Stack.
Scroll through the views. The points are fairly well-centered on this
highly reduced stack, even at high tilt. At this point, the best way to
see how well the erasing works is to run it.
- Press Erase Beads. When it is done, press View Erased Stack
to
to see the result. This erasing is probably adequate to avoid artifacts in
the tomogram, but there are steps you can take if you want to improve it.
You should see two points that have a crescent of bead left at the top even at
low tilt. These two points showed up as being particularly lower than the
beads in the previous
step of looking at the 2D model on the aligned stack.
- The easiest way to fix these problems is to press View 3D Model on Tomogram
again and find the two points in the tomogram. Click with the first mouse
button to select a point; this should bring up the nearest slice that the point
lies on. Zoom up and click with the third mouse button to center the point
better. Save the model and exit 3dmod.
- Press Reproject Model then Erase Beads. When done, press
View Erased Stack. The crescents should be gone at low tilt; you
may notice a few on other beads at high tilt. Another problem with the
erasing is that it leaves white halos. The only solution for these largely
aesthetic problems is to increase the Diameter to erase (pixels),
but the increase needs to be substantial, to 85 or 90.
- You do not need to press Use Erased Stack when doing 3D CTF correction.
2D Filtering for Dose Weighting:
Useful high-frequency information is progressively lost due to beam damage over
the course of tilt series acquisition. Dose-dependent attenuation of these
frequencies to reduce the residual noise is known as dose weighting.
- Switch to the 2D Filter tab.
- Check Apply dose weighting. This disables the simple
low-pass filter at the top of the dialog and enables the dose weighting options
below.
- The option Metadata in .mdoc file has been selected, and the
file "TS43.mrc.mdoc" exists, so you are all set to go on, but you should run the
filtering to see what it does.
- Press Filter. When it is done, press View Filtered Stack
and scroll through the views to see how the filtering increases at higher tilt.
- Again, there is no need to press Use Filtered Stack because
this filtering will be done as part of the 3D CTF reconstruction.
- Press Done to go on.
Tomogram generation:
At last, you can compute the tomogram. Notice that the "cryoAccurate.adoc"
template has selected Super-sample by 2, which will eliminate
most of the artifactual rays that would appear in an FFT of an XZ tomogram
slice; and it has set the Standard Gaussian cutoff to
0.425/pixel, higher than the usual 0.35, to preserve more of the high-frequency
information.
-
Select Erase Gold and Apply 2D filter.
-
Select Use the GPU if you have a GPU; the CTF correction will be much
faster. Select the number of processing units to use (cores or GPUs) in
the Parallel Processing table.
-
Compute slabs in parallel is appropriate if you have relatively
few processing units (e.g, 4 GPUs) or if there is plenty of storage space to
hold intermediate stacks for
all processing units at once.
-
Press Generate CTF-corrected Tomogram.
-
Press Use CTF-corrected Tomogram when it is done. We wil look at
the tomogram in the next step.
-
Press Done to go on.
Post-processing:
In this step, you can trim away unneeded regions, convert the tomogram to bytes
to save time and space, and reorient the tomogram so that the slices stored in
the file are in X/Y planes instead of X/Z planes. Even if you do not
want to trim or convert to bytes, you should always go through this step to get a
reoriented tomogram, which will work better with other
programs. Unfortunately, it is difficult see the VLPs well enough to
set reliable trimming limits, even if the tomogram is loaded with typical
amounts of binning. Here is a procedure that will work:
-
In the Trim vol panel of the Post-Processing
page, Right-click on 3dmod Full Volume and select Open
with startup window.
-
In the 3dmod startup window, set the Bin by spin boxes to
12 for both in X/Y and in Z.
-
Select Load non-byte data in as 16-bit integers; this will
preserve the dynamic range better when loading. Press OK.
-
The VLPs are visible in the Zap window, but if you scroll in Z you will see that
this view does not give a good definition of the tops and bottom limits of the
VLPs.
-
Open an XYZ window from the 3dmod Image menu or with the Ctrl-X hot key.
(The Slicer could be used instead). Set the Sum spin box
to 10.
- Turn on the rubber band in the Zap window but do not draw a rubber band.
You could do so to define an area to trim out in X and Y, but there is not much
area that could be trimmed here.
- In the XYZ window, scroll with the Y slicer to the low end until you see the
highest top of a VLP. Click with the first mouse button at a point well
above this top and press the Hi button in the Zap toolbar.
This should be about 10 pixels below the top in Z.
- Scroll to the high end in Y in the XYZ window and click a point below the
lowest end of a VLP. Press the Lo button in the Zap
toolbar.
- In Etomo, press Get XYZ Volume Range from 3dmod to import these limits,
which are now in unbinned pixels.
- Now we want to define a subvolume that will be used to determine scaling.
In the Zap window, scroll down to a Z of ~ 32 and draw a rubber band that excludes
the remnants of the gold fiducials. Press the first mouse button at the
lower-left corner and drag to the upper right corner to define the rubber band.
Select a narrower range of slices with Lo and Hi
that exclude the remnants of the gold when they show at higher Z.
- In Etomo, press Get XYZ from 3dmod in the Scaling section
to import these limits.
- Press Trim Volume. When done, press 3dmod Trimmed Volume
if you want to
see the result, but it is not very informative.
- Press Done to go on.
Making a Reduced, Filtered Volume
For particle selection, a much less noisy volume will be needed. A good
starting point is a volume reduced by 4, and filtered with the deconvolution
filter.
- Check Reduce by overall factor and enter 4 in the
text box. Leave the Factor in Z empty.
- Select the Deconvolution radio button.
- Enter 0.7 for the Deconvolution strength. The
Defocus entry can be left blank; the defocus will be taken from
the output of Ctfplotter.
- Press Reduce/Filter Volume. When it is done, press
Open Output Volume in 3dmod.
- Notice that the file is named by the reduction amount (which does not need to
be an integer).
- If the dynamic range of the densities is poor (with the black and white
sliders in 3dmod very close together), enter 2 in Data output
of mode
to make floating point output, and press Reduce/Filter Volume
again.
Clean Up:
In most cases, there is no need for the intermediate files from processing.
This step allows you to remove these files and leave all of the information from
which they could easily be recreated if necessary. The original raw tilt
series stack can also "archived" by compressing its difference from the current
stack; this operation is reversible.
- Press Archive Original Stack and confirm the deletion of the original
stack when it is done.
- All intermediate files are now shown in the box. Click in the box and
type Ctrl-A to select all files. You could then click on individual files
while holding down the Ctrl key to unselect them. You might wish to leave
cryo.preali (coarse aligned stack), cryo.ali (final aligned stack), or
cryo_full.rec (raw reconstruction, which could be trimmed differently).
- Press Delete Selected to remove the intermediate files that are still
selected.